Hyperglycemia, an abnormality that results from a breakdown of normal glucose control processes is also the result of molecular mechanisms to protect the insulin receptor? A hypothesis
Andrea Novas Maldonado1, Marina Martínez Sánchez2, Hugo Mendieta Zerón1,2
Abstract
Background: Deficiency in either ß-cell mass or function, or both, can lead to insufficient levels of insulin, resulting in hyperglycemia and diabetes mellitus.
Aim: This review raises the hypothesis that hyperglycemia is the result of cellular protective mechanisms of the insulin receptor.
Methodology: The methodology was a comprehensive review of existing literature on the insulin receptor and its response against molecular aggressors.
Results: Here we hypothesize that hyperglycemia is the result of a cellular mechanism of insulin receptor down-regulation to preserve its function, and at the same time, the ß-cell efficiency is diminished by the initial hyperinsulinemia, which is read as a negative feedback that, if it is long-lasting, becomes irreversible due to chronic apoptotic and dedifferentiation processes.
Conclusion: An increase in glucose plasma levels and its poor control triggers serious injuries caused by glucotoxicity and lipotoxicity, which affects mainly the pancreas but also leads to a systemic damage, triggering the activation of protective pathways attempting to preserve the homeostasis and prevent progression. In turn, these pathways produce an increase in glucose by decreasing the number of insulin cell receptors, thus avoiding deleterious effects on the cell.
Keywords: hyperglycemia, insulin receptor, Diabetes Mellitus.
Introduction
Type 2 Diabetes Mellitus (T2DM) is a chronic disease characterized by insulin resistance and damage to the pancreatic tissue that leads to chronic hyperglycemia, which in the long term ends up causing severe systemic damage, especially to blood vessels and nerves.1
The global T2DM prevalence in 2019 was estimated to be 9.3%, projected to reach 10.9% by 2045.2 Additionally, diabetes was the direct cause of death for 1.5 million people worldwide in 2019, with 48% of those deaths occurring before the age of 70. It is clear the association of this chronic disease with high carbohydrate diets, a boost in food availability, along with a sedentary lifestyle and changes in food nutritional content even in children, thus reducing the age of diagnosis and leading to a series of systemic metabolic consequences.3
The classical features of this syndrome include ß-cell insufficiency, insulin resistance, and hyperglycemia. This last one, in conjunction with inflammation, causes damage to pancreatic ß-cells, resulting in even less insulin production, raising glycemic levels and creating a vicious cycle of cell damage.4
Currently, almost all the literature on hyperglycemia in the onset of diabetes refers to this phenomenon as a sine qua non result of one of the damage mechanisms described in previous lines;5 however, the insulin receptor and the cellular cytoprotection mechanisms to preserve it against excessive blood glucose concentration have not been analyzed in the same depth and currently, cytoprotection mechanisms that were not previously known have been described.6
Mechanism of insulin action
Insulin is an endocrine peptide hormone composed of 51 aminoacids and is the most important anabolic hormone because it is necessary for the production, storage, and release of energy during the postprandial stage and fasting. It is produced by pancreatic ß-cells in response to increasing blood glucose levels, but it can also be stimulated by aminoacids and other hormones such as glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP).7
Insulin function consists not only of maintaining normal glucose blood levels by increasing glucose uptake in tissues, mostly muscle and liver, where it promotes its transformation into glycogen and triglycerides, but also is primarily responsible for controlling uptake, utilization, and storage of cellular nutrients, increases aminoacids and fatty acid uptake and storage; inhibits gluconeogenesis, glucogenolysis, and hepatic ketogenesis; promotes protein synthesis mainly at the muscle level; is antilipolytic; induces vascular relaxation in order to produce an increase in the subsequent utilization of glucose; and inhibits the secretion of glucagon by pancreatic alpha cells. All these actions are mediated through its binding to the cellular receptor, activating a series of intracellular signaling cascades.2,7,8
Insulin receptor
As insulin is a peptide hormone, its receptor is placed on the cellular surface. The insulin receptor (IR) is a heterotetrameric glycoprotein belonging to the family of growth factor receptors, which also have intrinsic tyrosine (Tyr) activity. Once insulin binds to the alpha subunit of the receptor, it causes conformational changes by activating the receptor, thereby initiating its catalytic activity.7,8,9
The insulin receptor is made up of two alpha subunits, which are found on the outside of the cellular membrane and are the site of insulin binding, and two beta subunits, which are divided into three sections: extracellular portion (the ectodomain, ECD), a single transmembrane helix and an intracellular tyrosine kinase domain.10 This intracellular zone is also divided into three structural regions:
- Juxtamembranal: an essential section for signal transmission; Tyr965 and Tyr972 are located here.
- A regulatory region: a place where the signal generated in the activating region is multiplied. Here we can find Tyr1158, Tyr1162, and Tyr1163.
- Activating region: where phosphorylation sites are located in the terminal carboxyl, which also functions as a signal regulator (Figure 1).7
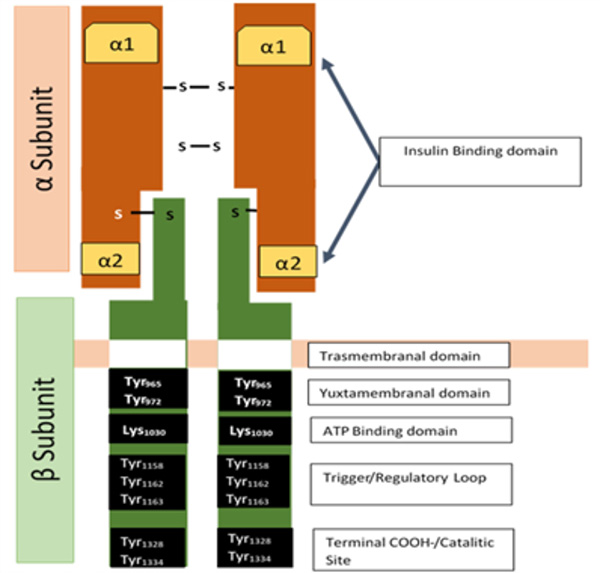
Figure 1. Insulin Receptor Structure
Insulin receptor is a heterodimeric transmembrane protein consisting in four subunits bind by disulfide bonds. Its subunits are composed of two a extracellular subunits, in which is located the hormone-binding domain in a1 and a2 regions; and two ß subunits, which has an extracellular domain, that connects with the a subunits, a transmembrane domain and a yuxtamembranal tyrosine kinase domain. Within its yuxtamembranal region are the ATP binding regions, activation/regulatory loop, and terminal carboxyl.
Once insulin binds to its receptor on the alpha subunits, the receptor activates, promoting autophosphorylation of tyrosine residues, which are consequently recognized by different adaptive proteins, including members of the insulin receptor substrate family (IRS-1 and IRS-2), which constitute the main substrates and common intermediates at the intracellular signal propagation stage. When adaptor proteins are coupled, intracellular signaling cascades are initiated. There are two main insulin signaling pathways; the phosphatidylinositol-3-kinase (PI3K) /Akt (protein kinase B (PKB)) pathway, which controls most metabolic actions; and the mitogen/Ras activated kinase (MAPK/Ras) pathway, related to regulation of gene expression and mitogenic effects.
Pathway of MAP Kinase Signaling
Phosphorylation of receptor subunits ß promotes its association with the Shc protein, which then binds to the Grb2/SOS complex. SOS acts as a recombinant factor of guanine nucleotides (GEEF), activating RAS and initiating MAP’s signaling. GTP-RAS activates RAF-1 by binding to it, resulting in the activation of MEK (MAP kinase kinase) and extracellular regulated kinase (ERK)-1 and 2. Once activated, ERK1/2 translocates to the nucleus, catalyzing the activation by phosphorylation of transcription factors, regulating genetic expression and promoting proliferation, growth, and cell differentiation (Figure 2).7,8,11
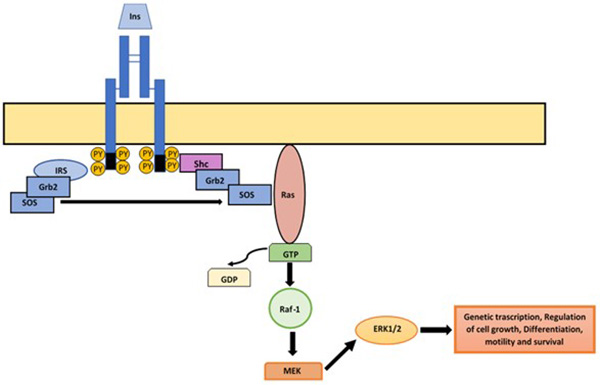
Figure 2. MAP kinase pathway
When insulin binds to its receptor, the MAPK pathway can be activated by two different pathways. In the first, the phosphorylation and activation of the receptor promotes the binding of Shc protein, to which the Grb2/SOS complex binds; SOS causes the switch from GDP to GTP consequently activating RAS. GTP-RAS joins and activates Raf-1, which will activate MEK and ERK1/2. Alternatively, there is Shc independent pathway where IRS-1 participates. In this, once activated the IR, the IRS-1 binds, linking to the Grb2/SOS complex following the same sequence as in the above described, once activated, ERK1/2 translocases to the nucleus provoking the phosphorylation of transcriptional factors involved on genetic transcription, cell growth, differentiation, motility and survival.
An alternative way to activate ERK1 and ERK2 is through the insulin receptor substrate (IRS), which is independent of Shc and binds directly to Grb2/SOS. Its activation results in actions related to protein synthesis and genetic expression in insulin-sensitive tissues but not in glucose transport regulation.7,8
The Phosphatidylinositol-3-Kinase Signaling Pathway
This pathway starts when the IR gets activated and phosphorylates along with IRS proteins, which interact with the IR activated receptor. Activated IRS proteins serve as binding and activation sites for other proteins with SH2 domains; p110 is transferred to the juxtamembrane zone, where it interacts with PI4-P and PI4,5-P2 substrates, which are phosphorylated to generate PIP2 and PIP3, which serve as binding sites for Ser kinases such as PDK1, AKT, and protein kinase B.7,9
Mtor/Rictor protein enzyme or PDK2 phosphorylates and activates AKT kinase in Ser43 and Thr 308 residues, promoting Thr phosphorylation by PDK1 by fully activating AKT.Glycogen kinase enzyme (GS), glycogen synthase kinase 3 (GSK3), inducible nitric oxide synthase (iNOS), phosphofructokinase 2 (PFK2), and cyclic AMP response element binding protein (CREB transcription factor), caspase 9 and the Bcl2 antagonist antiapoptotic protein (ADB) are among its substrates. This pathway is responsible for the metabolism of glucose, lipids, and synthesis in insulin-sensitive tissues (Figure 3).7
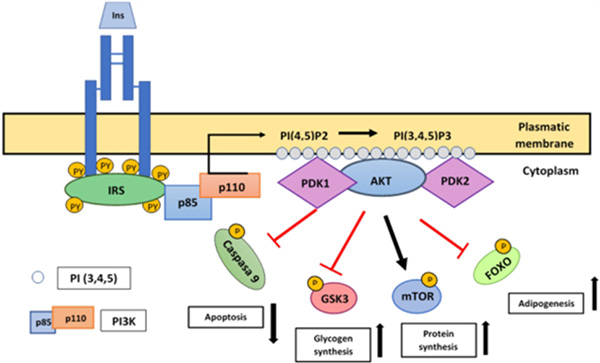
Figure 3. PI3K/AKT Pathway activation
When insulin binds to the IR and it gets active, it activates IRS, which contains several phosphorylation sites in Tyr residues, which when phosphorylated by IR, are transformed into binding and activation sites of proteins with SH2 domain as PI3K. PI3K is composed by a regulatory unit (p85) and a catalytic unit (p110). The interaction between p85/IRS-1 activates p110 and as a result, p110 has access to its PI(4,5)P2 substrate, which is phosphorylated at position 3 of inositol, generating PI(3,4,5)P3, serving as a binding site for Ser kinases such as PDK1 and Akt. The PDK2 protein complex activates Akt, inducing a first phosphorylation in Ser473 which is followed by a phosphorylation in Thr308, the latter induced by PDK1. This pathway is the main responsible of insulin metabolic action. Akt regulates many of the metabolic effects of insulin through the regulation of activation of various substrates that propagate the response, such as mTor, FOXO, GSK3 and caspase 9 acting over apoptosis, glycogen synthesis, protein synthesis and adipogenesis.
Glucose transport regulation
GLUT-4 mobilization to the plasmatic membrane in response to glucose in muscle and adipose tissue is dependent on the PI3K pathway and AKT kinase. Glucose metabolism at the hepatic level is promoted by glucokinase, which functions as a serum glucose sensor. Glucose metabolism results in an increase in the ATP/ADP ratio, leading to the closure of KATP channels and membrane depolarization, opening voltage-dependent calcium channels, which leads to the release of granules containing insulin (Figure 4).12
![]()
Figure 4. Glucose transport regulation
Insulin binding promotes the translocation of GLUT4 transporter from intracellular compartments to the plasma membrane. The translocation of Glut 4 can work in two different ways. In the first, the PI3K is involved. Once the IR is activated and phosphorylated, the IRS are phosphorylated and activated, recruiting effector molecules such as PI3K. The AS160 protein in its non-phosphorylated and active state negatively regulates the small G Rab proteins, which participate in the vesicular traffic of GLUT4. AS160 stimulates the hydrolysis of Rab-bound GTP, inactivating Rab, and inhibiting vesicular traffic. When AS160 is phosphorylated by Akt it is inhibited, so the Rab-dependent (active) traffic of GLUT4 to the plasma membrane is increased. The other pathway involves the proto-oncoprotein c-Cbl, in which insulin stimulates the formation of the c-Cbl - CAP complex to move to the surface by means of lipid rafts, the phosphorylation of c-Cbl recruits an adaptor protein called CrkII and C3G, an exchange factor for the GTPase TC10. TC10 is activated by C3G; TC10 participates in the activation of PKC-?/? that produces the translocation of GLUT4. Furthermore, PDK1 also causes phosphorylation of critical sites at the activation loop of two atypical forms of PKC (PKC?/?), which significantly contribute to insulin-induced GLUT4 translocation.
Insulin plays a crucial role in the regulation of GLUT-4, not only causing its translocation to the plasma membrane through exocytosis but also reducing endocytosis. Therefore, the rate of glucose transport into sensitive cells (fat and muscle) is controlled by the quantity of GLUT-4 on the cell surface and its duration on that site.9
Insulin resistance
Primary Mechanisms
Insulin resistance is initially seen as a decline in the first phase of insulin in response to glucose, followed by a progressive decrease in the second or late phase of glucose response.12 Chronic overconsumption, undernutrition, or cell stress can generate insulin resistance. Insulin resistance is a pathological state in which tissues normally sensitive to insulin effects cease to be so. In the genesis of insulin resistance, the glyco and lipotoxicity resulting from hyperglycemia and fat accumulation are responsible for the progression of the process.13
Excessive calorie intake would lead to the accumulation of liver fat as the liver constitutionally produces glucose in a postprandial state, only slowed down by the action of insulin. Insulin resistance caused by liver fat accumulation would cause an increase in postprandial glucose and an increase in insulin secretion to try to overcome resistance. Higher insulin levels encourage the conversion of carbohydrates to fat in the liver, creating a vicious circle that results in higher levels of serum insulin. The tendency to convert excess carbohydrates into fat increases the output of VLDL-triglycerides of hepatic origin, promoting the migration of ectopic fat acids and further damaging pancreatic cells until a point is reached where pancreatic cells cannot compensate for the increased demand for insulin.13 Importantly, this cycle can be reversed if excess carbohydrates are resolved, and the fat load is removed from the organs. Furthermore, this cycle is reinforced by a decreased oxidation of fatty acids and lipolysis secondary to impaired sympathetic system activation in white and brown cells.3,13,14
Likewise, a very important step of the insulin resistance mechanism is the regulatory mechanisms at the insulin receptor level, as in the modification of the signaling pathway. Within these regulatory mechanisms, one of the most closely related is the increased phosphorylation of Ser/Thr at the IR level, which alters its association with key proteins in the pathway, blocks Tyr activation sites and induces their degradation. This is followed by the secretion of leptin hormone, which generates chronic inflammation, provoking counterinflammation, being overcome by the action of insulin by the need to store energy.2
ß-cell glucotoxicity
The theory of cellular damage by glucotoxicity was initiated when it was realized that patients, especially those who had poor glycemic control, experienced a gradual deterioration in their metabolic control even with the use of more than two antidiabetic drugs.4 At diagnosis, it is estimated that there is already a 50% decrease in the function of the ß-cells which continues to decline over time, whereas, after 10 years, 50% of patients will require insulin therapy. ß-cells apoptosis plays an important role in the development and progression of diabetes mellitus and the subsequent need for insulin therapy.4,12,15
Bolaffi et al. showed that prolonged stimulation with glucose results in a reduced insulin secretory response.16 The glycosylation of cellular proteins induced by chronic exposure to high levels of glucose is a critical element in cell dysfunction. An islet grown for a time of only 48 hours in high concentrations of glucose (16.7 mmol/L) showed a 70-90% decrease in response to glucose in relation to cells grown at low glucose levels, showing a desensitization towards further stimuli, which led to the hypothesis of a regulatory mechanism against a hyperosmolar environment.10,16
On the other hand, due to the high glucose concentration, the reactive oxygen species creation within the ß-cell increases. Added to this, the islets have intrinsically low antioxidant enzymes to deal with that increase. This has been supported by studies where it is found markers of oxidative stress and elevated pro-oxidants in the serum of diabetics compared to non-diabetics, which decreased along with better glycemic control, insulin secretion, and oral glucose tolerance test.4
At a transcriptional level by means of oxygen-free radicals, hyperglucemia affects insulin sensitivity by reducing both DNA binding activity and mRNA levels of PDX-1, the nuclear factor responsible for insulin trans-activation; increasing the activity of C/???ß, a negative regulator of the genetic transcription of insulin in ß cells; and reducing the levels of RIP RIPE3bl-Act, a positive regulator of insulin expression. All these changes can be prevented by adding aminoguanidine or V-acetylcysteine (antioxidants), confirming free oxygen radicals as the genesis of these alterations.12
To sum up, all these processes secondary to free radicals lead to impairment of ß-cell function, resulting in increased basal insulin release, reduced response to stimulus to secrete insulin, and a gradual depletion of insulin stores. In addition, it was found that although in normal conditions, glucose acts as a powerful cell growth enhancer, in cases of chronic hyperglycemia, was shown to reduce ß-cell proliferation and increase apoptosis.12
Another key factor in glucotoxicity is AMPK, which is called the master regulator of energy metabolism, involved in normoglycemia and glucose homeostasis and whose activation is regulated by energy levels. Inflammation generated by obesity suppresses AMPK, causing glucolipotoxicity and therefore insulin resistance.14 Although lipotoxicity plays a role in damaging the ß-cell, it only occurs in the context of hyperglycemia. However, glucose toxicity can occur even in the absence of hyperlipidemia. Furthermore, most type 2 diabetes patients are obese, and their lipid levels in the blood worsen as glycaemia becomes higher, accumulating in ß-cells and causing further damage.
Lipotoxicity
Although fatty acids serve as a fuel for pancreatic ß-cells under physiological conditions, chronic exposure is toxic, causing decreased insulin secretion, genetic expression, and increased cell death. Mitochondrial oxidation generally eliminates unnecessary fatty acids at normal glucose concentrations. However, when glucose levels are elevated, there is a pathological accumulation that impairs ß-cell function and peripheral tissue. In addition, it interferes with insulin signaling by activating serine phosphorylation in IRS-1.14
Inflammation as a regulator of insulin response
Low-grade inflammation, secondary to obesity, leads to modifications in adipose, hepatic, and muscular tissue. Thus, alteration in the adipokine secretion, increased production of inflammatory cytokines, as well as FFA release act on hepatic and muscular cells, impairing their response to insulin.8,17 Some of the mechanisms by which inflammatory cytokines promote insulin resistance are: activation of Ser/Thr kinases, decreased expression of IRS-1, or expression and activation of SOCS-3. In addition, changes produced by obesity activate toll-like receptors (TLR), in particular TLR-2 and TLR-4.7,8,18 This last is induced by toxic blood levels of glucose and FFA, cytokine signaling, and ER stress.
Obesity increases inflammatory cytokine secretion, such as JNK1, MyD88, and NF-kB, which contributes to insulin resistance and increases the expression of enzymes involved in biosceramide synthesis. This activates PP2A phosphatase, which dephosphorizes Akt, inhibiting the insulin response and promoting ß-cell apoptosis.8,14 Besides, Tumor Necrosis Factor alpha (TNF-a) plays an important role in pathogenesis by causing lipolysis and serine phosphorylation of IRS proteins, thereby blocking downstream signaling while promoting cytokine secretion. It has been proven that because of removing TNF, secondary to low-grade inflammatory response, it improves insulin sensitivity in obese mice.3,8,14,19
Correspondingly, an excess of glucose induces up-regulation of FAS receptors,15 leading to activation of caspases-8 and 3, resulting in DNA fragmentation by binding to their ligand in neighboring cells. Other findings, using human cells, found that when cultured with high concentrations of glucose, they modified their protein pattern and expression, increasing pro-apoptotic and decreasing anti apoptotic.12,19
Mitochondrial dysfunction
An increase in FFA concentration due to obesity produces a mitochondrial overload and an increase in ß-oxidation in muscle, liver, and brown fat in order to increase the bioavailability of energy, producing an exaggerated amount of ATP. When ATP exceeds a threshold, the excess energy produces a substrate inhibition in mitochondrial function.8
ATP inactivates the AMP-activated protein kinase (adenosine monophosphate) enzyme to reduce insulin-induced glucose uptake and decrease ATP production. Insulin resistance thus acts as a cellular protective mechanism that aims to control the ATP-induced stress response in muscle and liver. The decrease in oxidation of substrates affects the flow of electrons to the respiratory chain, causing its union with oxygen, forming superoxide ions, which in turn cause mitochondrial damage, protein aggregation, and lipid peroxidation, leading to mitophagy, apoptosis, impaired release of insulin by ß-cells and Ser/Thr kinase activation.8
Downstream regulatory mechanisms from the IRS
PI3K activation product inhibition
Proteins such as inositol phosphatase with SH2 domain (SHIP2) or phosphatase and tensin homologue removed on chromosome 10 (PTEN) act as lipid phosphatases, dephosphorifying PIP3, antagonizing the PI3K/Akt signal and dephosphorifying IRLS-1, important in downstream signaling of insulin. By inhibiting these proteins, an increase in insulin sensitivity is seen by increased action and production of PIP3.2,7,9
Insulin action regulation at the molecular level
In the presence of insulin, AKT kinase phosphorylates the transcriptional factor FoxO1, causing its interaction with the protein 13-3-3, causing its release from the cell nucleus and subsequent proteosomal degradation, preventing the transcription of the insulin gene.8
A number of extrinsic factors that have been seen to be related to insulin resistance include TNF-a, interleukins (IL) (IL-6, IL-18, IL-1)14 catabolic hormones such as epinephrine, glucagon, and angiotensin II, endothelin, and even locally produced hormones such as resistin (adipose tissue), have a direct relationship with fatty acids, such as triglycerides and phospholipids, which are able to inhibit the PI3K enzyme, increase phosphorylation of Ser/Thr residues in IRS-1, and alter the AKT activation pathway by increasing the amount of ceramide and diacylglycerol in muscle tissue.5,8,11,17 Intrinsic cellular factors include mitochondrial dysfunction, oxidative stress, and plasma reticulum stress. Hyperinsulinemia worsens IRS-1 Ser/Thr phosphorylation by activating the PI3K/AKT, PKC-/-, or mTORC1/p70S6k insulin signaling pathways.8
Mechanisms of insulin receptor regulation
Endocytosis
Once insulin binds to its receptor, the insulin receptor complex is internalized into endosomes by means of clathrin-coated vesicles that are invaginated, the receptor being still active and phosphorylated. Within the endosomes, the acid Ph dissociates the complex and insulin is degraded by the endosomal acid insulinase enzyme, while the receptor is recycled back to the surface. However, under conditions of hyperstimulation, the IR undergoes degradation in the lysosomes. This is a crucial process that determines the number of receptors present on the cell surface and is crucial for insulin sensitivity, modifying the glucose rate of transport into the cells (Figure 5).8,9
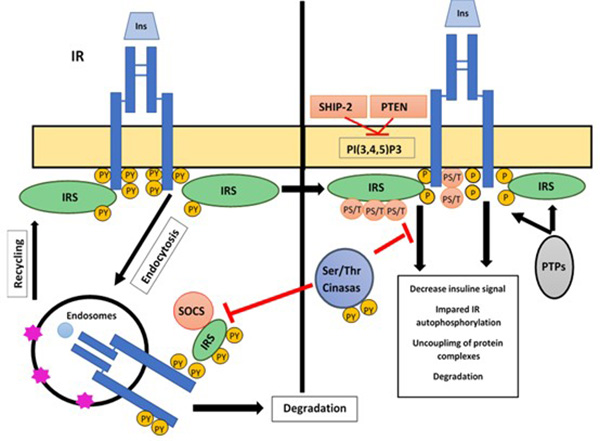
Figure 5. Insulin signal regulation mechanisms
Actions of insulin are modulated through different mechanisms including: a) endocytosis and recycling of receptors, which control their degradation and number in the cell membrane through a negative regulatory mechanism; b) the action of proteins with tyrosine phosphatase activity (PTPs), which dephosphorize key protein residues from insulin signaling such as the IRS and its own receptor, and c) phosphorylation in IR and IRS Ser/Thr residues. These mechanisms regulate the insulin signal at a receptor level or proteins downstream of it, impairing its activity, decoupling the formation of protein complexes and regulating its number and cell location.
Phosphatases
This mechanism of action includes the deposphorylation of key Tyr residues in the activation of receptor subunits B by the activation of Tyr phosphatases. The action of these phosphatases (PTPs) causes a decrease in the incorporation of glucose into muscle and adipose tissue and metabolic alterations. Within them is phosphotyrosine phosphatase 1B (PTP-1B); in comparison, there are proteins such as SHP-2, which have a dual effect, both negative and positive; negative binding to IR and IRS-1 inactivates them by defibrillation and positive in the Ras/MAPK pathway (Figure 5).7-9
Ser/Thr residues phosphorylation
In response to prolonged insulin stimulation, there is an increase in phosphorylation in Ser/Thr residues in the IR, impairing its response to insulin and resulting in decoupling in the pathway.7,8 This decoupling has four fundamental mechanisms, which are: a) dissociation of pathway effector proteins and IR, affecting their ability to phosphorylate and attenuate signaling; b) degradation of IRS-protein 1 or conversion to IR kinase activity inhibitors. Due to its modulatory function in the juxtamembranal and catalytic regions, PKC has been implicated as a key kinase in this regulatory model due to its modulatory function in the juxtamembranal and catalytic regions.2,8,14,17,19
Another example of molecules that can trigger this effect in IRS-1 are SOCS proteins, a family of cytokine signaling protein suppressors. Insulin action in several tissues causes the expression of SOCS-1 and SOCS-3 proteins, which inhibit the insulin receptor in various ways: a) inhibiting the phosphorylation in receptor tyrosines by competing for the same site of interaction; b) promoting IRS proteasomal degradation; and c) inhibiting IR kinase activity (Figure 5).7,8,17,19
Diabetes reversibility: evidence of insulin receptor recovery
Current studies indicate that diabetes mellitus is a condition based on fat accumulation in the liver and pancreas, causing lipo and glucotoxicity;20 yet weight reduction, either by surgical or dietary methods, can normalize hepatic response to insulin, ß-cell recovery, and normalization of glucose levels in early diagnosis.21
Within the primary care-based Diabetes Remission Clinical Trial (DiRECT), it was shown that 46% of people were able to get diabetes remission at 12 months and 36% at 24 months by weight loss, showing the dependence of the time of evolution of the disease with the success rates.13 Considering the pathophysiological mechanism of insulin resistance and its relationship with excess fat and carbohydrates, the role of bariatric surgery and caloric-restricted diet on the reversibility of diabetes mellitus has been investigated.22
In either case, during a caloric restriction, the body would have to start using stored energy reserves, causing accumulated fat in the liver and pancreas to decrease, improving insulin resistance and thus lowering fasting glucose levels, as well as improving pancreatic ß-cell function and restoring insulin secretion.13 Bauman et al. implemented a low-calorie diet of 900 kcal, including 115g of protein, which led to a significant improvement in glycemic control that was mainly attributed to improvements in insulin sensitivity.23
One study showed that patients with LCD and gastric bypass had equally beneficial levels of improvement in a short time.24 However, this remission was not as long as the bypass, with few reaching remissions at the age of follow-up. There was a decrease in the need for hypoglycemic medication. The caloric level necessary to achieve remission has not been standardized. Some authors have obtained satisfactory results with diets of 600 kcal/day and 825-853 kcal/day, showing better markers with a shorter evolution time of diabetes.15
The Direct25 and Counterbalance26 studies showed consistent results in which remission was achieved by keeping diabetes free while avoiding weight regain.12 According to the counterbalance study, the patients who benefited from caloric restriction were those who had an evolution time of less than 11 years since the diagnosis of diabetes, because over time, damage to the ß-cell becomes irreparable.13
Bariatric surgery is currently indicated for patients with a BMI greater than 40, or >35 kg/m2 with comorbidities). The change in insulin sensitivity occurs in hours or days, which is very likely to represent changes at the endocrine and cell signaling level. After a Roux-en-Y Gastric Bypass (RYGB), there is a weight loss on average of 25-30% of total body weight, maintained for at least 10 years; in fact, within a few hours there is an improvement in plasma glucose levels, reduction of the necessary dose in hypoglycemic medication and a reversibility of up to 80% of patients, showing its improvement on pancreatic ß-cells sensitivity and function. The effectiveness of the RYGB depends on the severity of the disease and ß-cell function.9,12,14,26
It has been shown that fat, obese, and type 2 diabetic patients exhibit higher lipid oxidation in insulin-stimulated surroundings instead of normally turning their metabolism against glucose oxidation.2,4,8,11,18
Among the changes that have been seen after surgery are an improvement in the endocrine profile modulating euglycemia; changes in the intestinal microbiota with reversal of the obesogenic phenotype; and improvement in fatty acid metabolism.11,15 Within one week after the surgery, patients experienced improved liver fasting insulin clearance, reduced de novo glucose production basal, and increased hepatic insulin sensitivity. At 3 months, improved pancreatic ß-cell functionality and glucose sensitivity increased release of GLP-1, PYY, and other anorexigenic hormones such as oxyntomodulin and cholecystokinin, which may contribute to weight loss and increased insulin sensitivity in muscle and adipose tissue.14,27
Once the mechanism of activation and damage of the insulin receptor has been reviewed, with the clear evidence of reversibility to an efficient state of response after bariatric surgery or a strict diet, it is clear that in an initial stage of damage, the response is the protection of the insulin receptor, but as occurs in different hormonal axes, when these are inhibited for a long period, they remain atrophied forever.28
2. Conclusion
To sum up, it has been proved that an increase in glucose plasma levels and its poor control triggers serious injuries caused by glucotoxicity and lipotoxicity, which affects at both a pancreatic and systemic level, triggering the activation of pathways that attempt to compensate for damage and maintain homeostasis to prevent progression.29 In turn, these pathways produce an increase in glucose by decreasing the number of insulin cell receptors, thus avoiding deleterious effects on the cell.
On the other hand, it has been observed that good glycemic control with low carbohydrate diets or secondary to bariatric surgeries can have a positive effect in a few weeks because of changes in glucose metabolism, causing a decrease in cell damage and restoring homeostasis.30 Our hypothesis is that it would consequently lead to the replacement of insulin cell receptors and thereby the clinical remission of diabetes
References
- Nellaiappan K, Preeti K, Khatri DK, Singh SB. Diabetic Complications: An Update on Pathobiology and Therapeutic Strategies. Curr Diabetes Rev. 2022;18(1):e030821192146.
- Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA, Ogurtsova K, Shaw JE, Bright D, Williams R; IDF Diabetes Atlas Committee. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. 2019 Nov;157:107843.
- Saltiel AR. Insulin resistance in the defense against obesity. Cell Metab. 2012;15(6):798-804.
- Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004;53 Suppl 1:S119-24.
- Morgan NG. Fatty acids and beta-cell toxicity. Curr Opin Clin Nutr Metab Care. 2009;12(2):117-22.
- Pietrocola F, Izzo V, Niso-Santano M, Vacchelli E, Galluzzi L, Maiuri MC, Kroemer G. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol. 2013;23(5):310-22.
- Olivares Reyes JA, Arellano Plancarte A. Bases Moleculares de las Acciones de la Insulina. REB Rev Educ Bioquímica. 2008;27(1):9-18.
- Gutiérrez-Rodelo C, Roura-Guiberna A, Olivares-Reyes JA. Mecanismos Moleculares de la Resistencia a la Insulina: Una Actualización [Molecular Mechanisms of Insulin Resistance: An Update]. Gac Med Mex. 2017;153(2):214-228.
- Chang L, Chiang SH, Saltiel AR. Insulin signaling and the regulation of glucose transport. Mol Med. 2004;10(7-12):65-71.
- Scapin G, Dandey VP, Zhang Z, Prosise W, Hruza A, Kelly T, Mayhood T, Strickland C, Potter CS, Carragher B. Structure of the insulin receptor-insulin complex by single-particle cryo-EM analysis. Nature. 2018;556(7699):122-125.
- Rosa G, Mingrone G, Manco M, Euthine V, Gniuli D, Calvani R, et al. Molecular mechanisms of diabetes reversibility after bariatric surgery. Int J Obes (Lond). 2007;31(9):1429-36.
- Bergman M. Pathophysiology of prediabetes and treatment implications for the prevention of type 2 diabetes mellitus. Endocrine. 2013;43(3):504-13.
- Yang X, He Z, Chen Q, Chen Y, Chen G, Liu C. Global research trends of diabetes remission: a bibliometric study. Front Endocrinol (Lausanne). 2023;14:1272651.
- Alipourfard I, Datukishvili N, Mikeladze D. TNF-a Downregulation Modifies Insulin Receptor Substrate 1 (IRS-1) in Metabolic Signaling of Diabetic Insulin-Resistant Hepatocytes. Mediators Inflamm. 2019;2019:3560819.
- Hallberg SJ, Gershuni VM, Hazbun TL, Athinarayanan SJ. Reversing Type 2 Diabetes: A Narrative Review of the Evidence. Nutrients. 2019;11(4):766.
- Bolaffi JL, Heldt A, Lewis LD, Grodsky GM. The third phase of in vitro insulin secretion. Evidence for glucose insensitivity. Diabetes. 1986;35(3):370-3.
- Rehman K, Akash MS. Mechanisms of inflammatory responses and development of insulin resistance: how are they interlinked? J Biomed Sci. 2016;23(1):87.
- Shyr ZA, Wang Z, York NW, Nichols CG, Remedi MS. The role of membrane excitability in pancreatic ß-cell glucotoxicity. Sci Rep. 2019;9(1):6952.
1 - Jiang Y, Pagadala J, Miller D, Steinle JJ. Reduced insulin receptor signaling in retinal Müller cells cultured in high glucose. Mol Vis. 2013;19:804-11.
- Taylor R, Al-Mrabeh A, Sattar N. Understanding the mechanisms of reversal of type 2 diabetes. Lancet Diabetes Endocrinol. 2019;7(9):726-736.
- Wolfe BM, Kvach E, Eckel RH. Treatment of Obesity: Weight Loss and Bariatric Surgery. Circ Res. 2016;118(11):1844-1855.
- Almaghrbi R, Alyamani R, Aliwi L, Moawad J, Hussain A, Wang Y, Shi Z. Association between Dietary Pattern, Weight Loss, and Diabetes among Adults with a History of Bariatric Surgery: Results from the Qatar Biobank Study. Nutrients. 2024;16(14):2194.
- Bauman WA, Schwartz E, Rose HG, Eisenstein HN, Johnson DW. Early and long-term effects of acute caloric deprivation in obese diabetic patients. Am J Med. 1988;85(1):38-46.
- Jackness C, Karmally W, Febres G, Conwell IM, Ahmed L, Bessler M, McMahon DJ, Korner J. Very low-calorie diet mimics the early beneficial effect of Roux-en-Y gastric bypass on insulin sensitivity and ß-cell Function in type 2 diabetic patients. Diabetes. 2013;62(9):3027-3032.
- Lean ME, Leslie WS, Barnes AC, Brosnahan N, Thom G, McCombie LC Primary care-led weight management for remission of type 2 diabetes (DiRECT): an open-label, cluster-randomised trial. Lancet. 2018;391(10120):541-551.
- Wei J, Chen J, Wei X, Xiang X, Cheng Q, Xu J, et al. Long-term remission of type 2 diabetes after very-low-calorie restriction and related predictors. Front Endocrinol (Lausanne). 2022;13:968239.
- Pop LM, Mari A, Zhao TJ, Mitchell L, Burgess S, Li X, et al. Roux-en-Y gastric bypass compared with equivalent diet restriction: Mechanistic insights into diabetes remission. Diabetes Obes Metab. 2018;20(7):1710-1721.
- Mendieta Zerón H, Hinojosa Juárez A, Vargas Hernández J. Stem cells in endocrinology. Facts and future. In: Atta-ur-Rahman and Shazia Anjum (Eds.). Frontiers in Stem Cell and Regenerative Medicine Research. Volume 8. Bentham Science Publishers. 2018, pp. 150-167. DOI: 10.2174/9781681085890118080006.
- van Raalte DH, Verchere CB. Improving glycaemic control in type 2 diabetes: Stimulate insulin secretion or provide beta-cell rest? Diabetes Obes Metab. 2017;19(9):1205-1213.
- Sayyed Kassem L, Rajpal A, Barreiro MV, Ismail-Beigi F. Beta-cell function in type 2 diabetes (T2DM): Can it be preserved or enhanced? J Diabetes. 2023;15(10):817-837.